The Microbial Kinome Switch: How TMA Inhibition of IRAK4 Rewrites Metabolic Health
December 8, 2025
December 3, 2025
Obesity is a global health crisis characterized by excessive adipose tissue accumulation and chronic low-grade inflammation, which serves as a precursor to type 2 diabetes and cardiovascular diseases. This study, titled "Adipocytes orchestrate obesity-related chronic inflammation through β2-microglobulin," published in Signal Transduction and Targeted Therapy (2025), addresses a critical yet poorly understood mechanism: how adipocytes directly communicate with the immune system. While past research focused heavily on how immune cells spontaneously migrate into fat tissue, the specific molecular "bridges" used by hypertrophic adipocytes to initiate this inflammatory cascade remained elusive. The researchers identified that β2-microglobulin (B2M), traditionally known for its role in the immune system, is the missing link that converts metabolic stress into a full-blown inflammatory response.
The research team mapped the role of B2M through sophisticated genetic models and multi-omics analysis, revealing a complex network where B2M acts as a master switch for metabolic inflammation.
Transcriptome analysis of mature adipocytes from mice fed a high-fat diet (HFD) revealed that antigen processing and iron transport pathways were the most significantly upregulated systems. B2M emerged as the central node connecting these two pathways. Validation experiments confirmed that B2M expression is specifically elevated in hypertrophic adipocytes, while other metabolic organs like the liver remain initially unaffected.
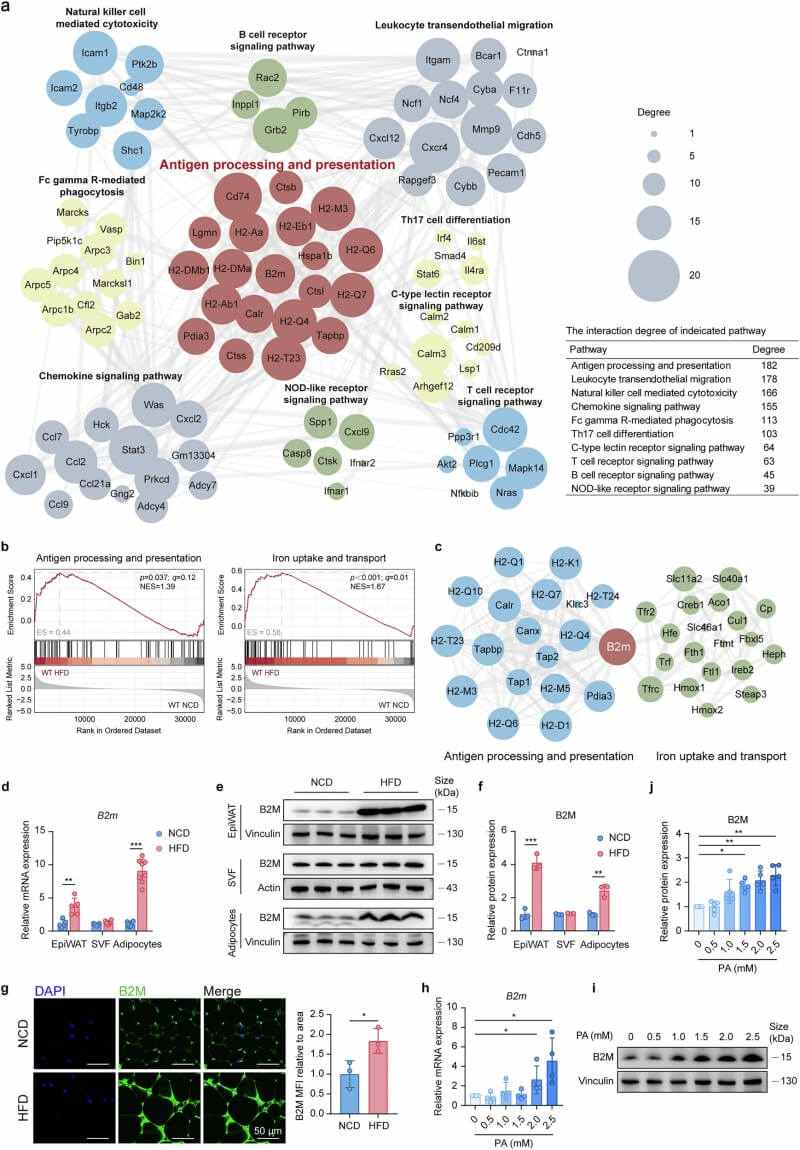 Fig.1 B2M is highly expressed in hypertrophic adipocytes and increases with HFD feeding or palmitic acid treatment. (Li, et al., 2025)
Fig.1 B2M is highly expressed in hypertrophic adipocytes and increases with HFD feeding or palmitic acid treatment. (Li, et al., 2025)
The researchers generated adipocyte-specific B2M knockout (B2mcKO) mice by crossing B2mf/f mice with Adipoq-Cre mice. When challenged with an HFD, these mice were remarkably resistant to weight gain, exhibited smaller adipocyte size, and maintained superior glucose tolerance and insulin sensitivity. Crucially, the absence of B2M reduced the infiltration of inflammatory CD8+ T cells and M1-polarized macrophages by nearly 70%.
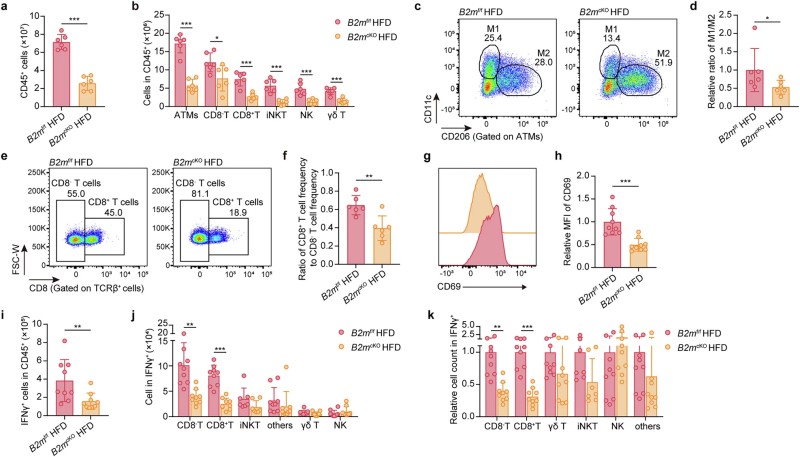 Fig.2 Deleting B2M in adipocytes reduces HFD-induced infiltration of inflammatory immune cells in adipose tissue. (Li, et al., 2025)
Fig.2 Deleting B2M in adipocytes reduces HFD-induced infiltration of inflammatory immune cells in adipose tissue. (Li, et al., 2025)
The study found that B2M is essential for the stable membrane localization of MHC-I in adipocytes. In obesity, adipocytes increase MHC-I presentation, effectively acting as APCs. Co-culture assays demonstrated that hypertrophic adipocytes directly activate adipose-resident CD8+ T cells in a B2M- and cell-contact-dependent manner, a process that bypasses soluble factors.
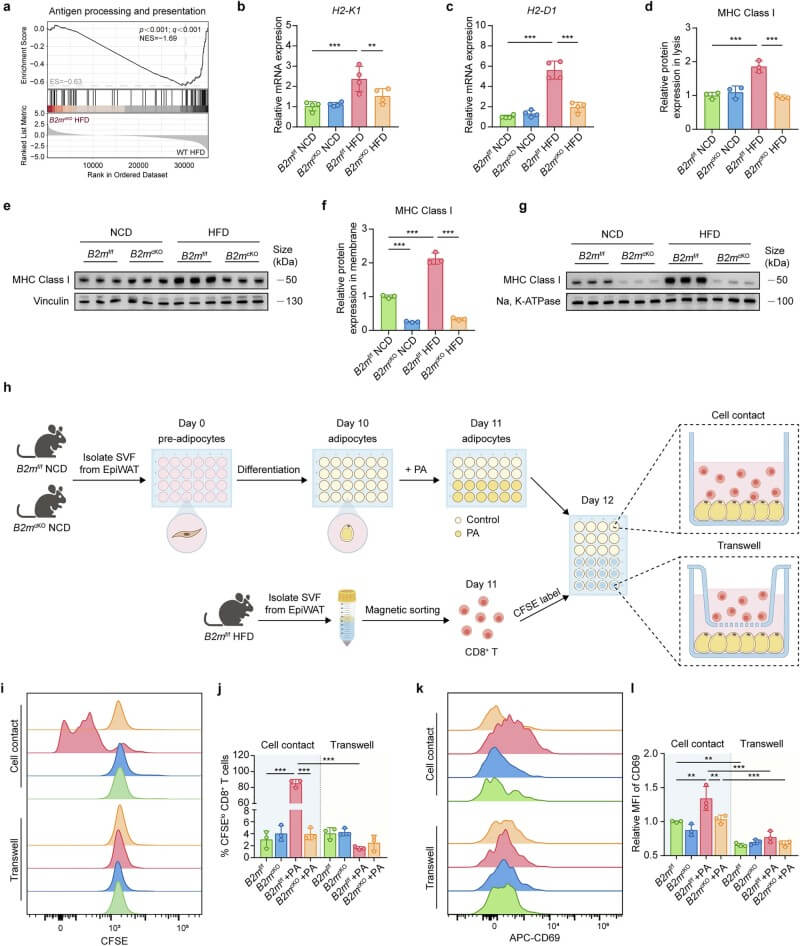 Fig.3 Activation of adipose-resident CD8+ T cells by hypertrophic adipocytes depends on B2M and direct cell contact. (Li, et al., 2025)
Fig.3 Activation of adipose-resident CD8+ T cells by hypertrophic adipocytes depends on B2M and direct cell contact. (Li, et al., 2025)
B2M was found to govern iron flux through the HFE/B2M-TFR1/2-hepcidin-FPN axis. In obese adipocytes, HFE shifts its binding from TFR1 to TFR2, a move facilitated by B2M. This shift triggers hepcidin expression, which degrades the iron exporter FPN, leading to massive intracellular iron overload. Notably, this iron dysregulation in fat occurs much earlier than in the liver, suggesting adipose tissue is more sensitive to metabolic iron stress.
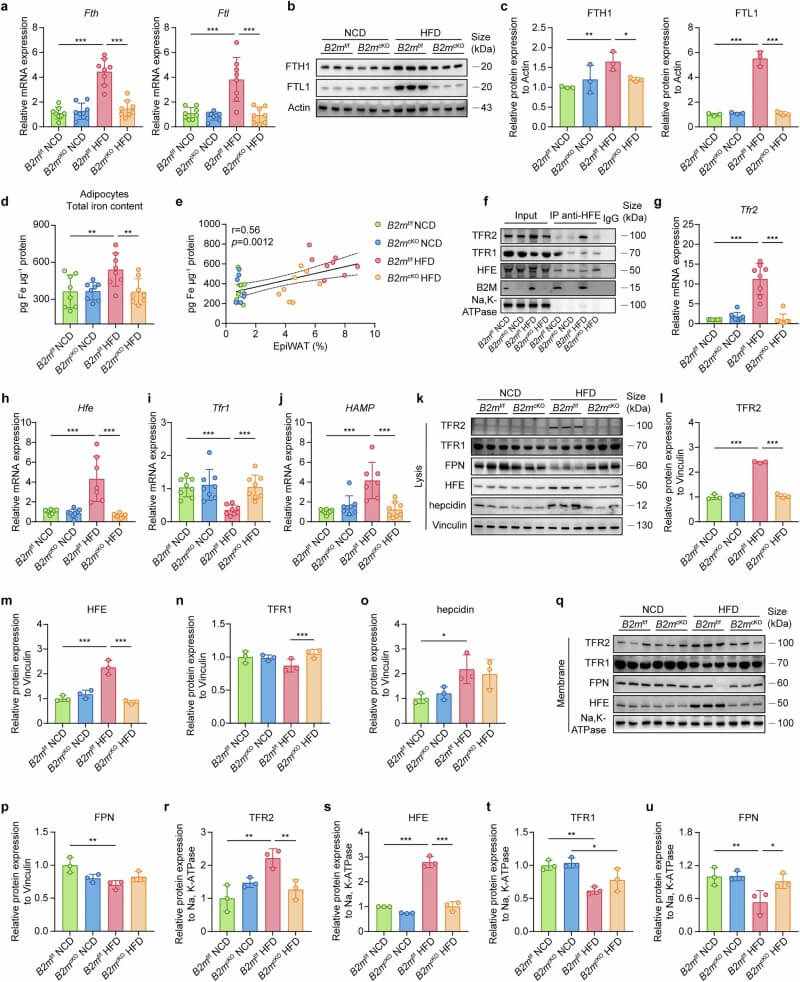 Fig.4 B2M deficiency prevents HFD-induced iron accumulation in adipocytes and alters iron-related protein expression. (Li, et al., 2025)
Fig.4 B2M deficiency prevents HFD-induced iron accumulation in adipocytes and alters iron-related protein expression. (Li, et al., 2025)
The resulting iron overload triggers the Fenton reaction, producing toxic ROS and lipid peroxidation—the hallmarks of ferroptosis. The team observed that ferroptotic adipocytes are encircled by macrophages in "crown-like structures". While ferroptosis is not required for T-cell activation, it is essential for M1 macrophage polarization; inhibiting ferroptosis with Ferrostatin-1 effectively blocked the inflammatory shift in macrophages.
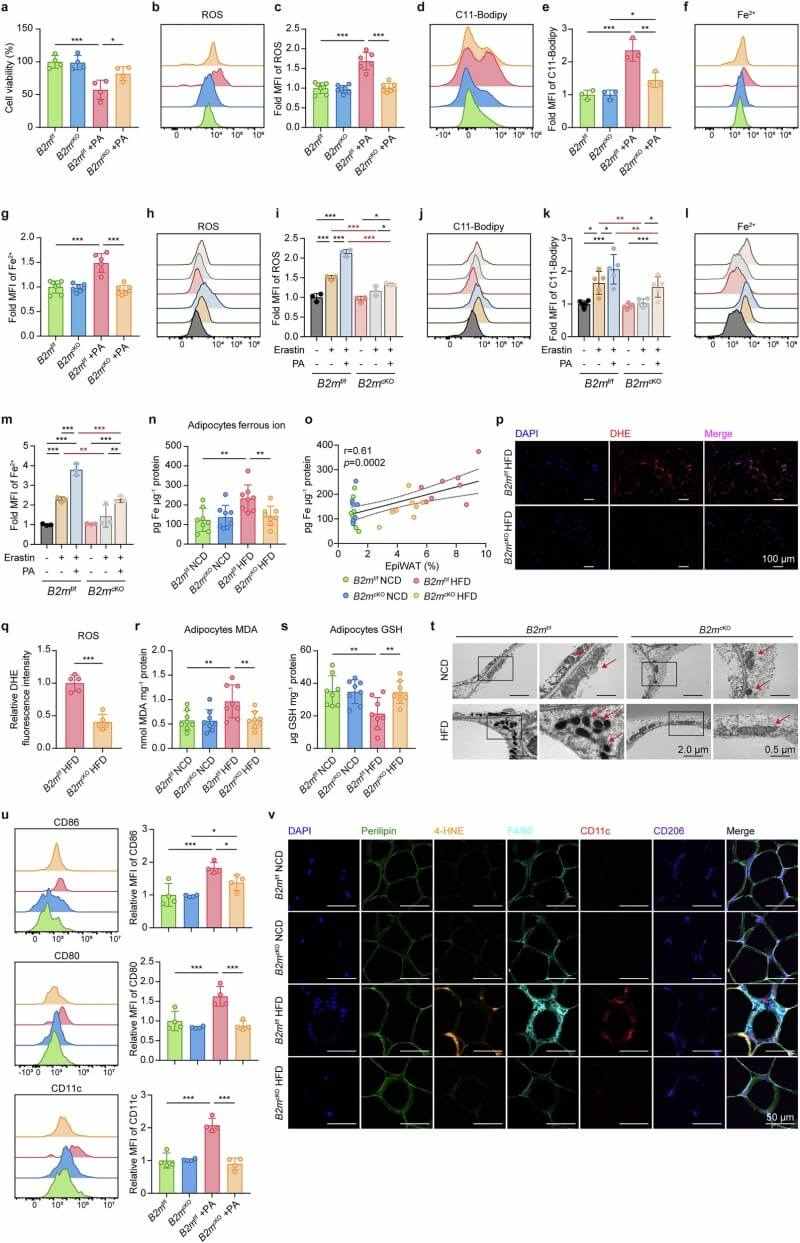 Fig.5 Lack of B2M alleviates HFD-induced adipocyte ferroptosis and reduces M1 macrophage polarization. (Li, et al., 2025)
Fig.5 Lack of B2M alleviates HFD-induced adipocyte ferroptosis and reduces M1 macrophage polarization. (Li, et al., 2025)
To assess therapeutic potential, the team injected AAV9 vectors carrying B2m-shRNA directly into the bilateral epididymal white adipose tissue (EpiWAT) of obese mice. This targeted intervention successfully reduced B2M in fat, reversed weight gain, and restored metabolic health. Finally, human data from subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) confirmed these findings: B2M expression in human visceral fat is significantly higher in obese individuals and correlates positively with BMI and insulin resistance.
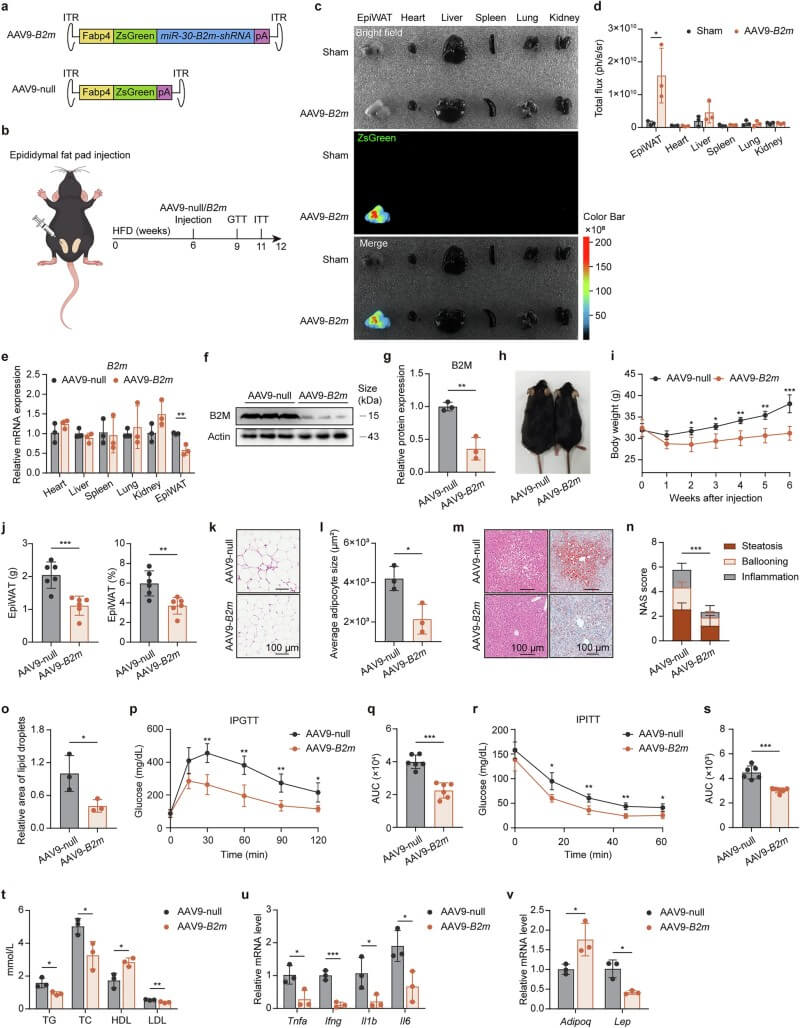 Fig.6 AAV-mediated B2M knockdown in adipose tissue improves obesity, metabolic parameters, and inflammation in HFD-fed mice. (Li, et al., 2025)
Fig.6 AAV-mediated B2M knockdown in adipose tissue improves obesity, metabolic parameters, and inflammation in HFD-fed mice. (Li, et al., 2025)
This landmark study redefines the adipocyte as a central "orchestrator" of the immune microenvironment. By identifying the B2M-dependent dual pathways—antigen presentation to T cells and iron-driven ferroptosis for macrophage polarization—the researchers have provided a unified model for obesity-related inflammation.
However, the study also acknowledges that B2M knockout does not completely eliminate adipocyte death. This suggests the existence of B2M-independent mechanisms, such as lipotoxicity-induced oxidative stress, which contribute to "residual" cell death. Future research must now determine the relative contributions of these parallel pathways to chronic inflammation.
The translational significance is profound: since B2M levels are elevated in human obesity and its targeted knockdown in mice yields dramatic therapeutic benefits, B2M serves as a high-priority target for future drug development. This provides a foundation for developing specific adipose B2M inhibitors to treat obesity and related metabolic disorders at their source.
For researchers dedicated to unraveling the complex physiological and pathological mechanisms of adipose tissue biology, ferroptosis, or metabolic immunology, Protheragen offers a comprehensive suite of high-precision preclinical solutions. We provide integrated, end-to-end specialized services designed to accelerate your mechanistic studies and therapeutic discovery.
Reference
Copyright © Protheragen. All rights reserves.